
SeqFu toolkit
@SeqFu2
Mingling sequences in Fasta and Fastq formats ~ Plus other #bioinformatics tips for NGS analyses from the command line
You might like







SeqFu 1.22.0 is out, with some minor improvements and updates. It's codenamed #EBAME9 because it features a new `--anvio` mode in "seqfu cat" github.com/telatin/seqfu2…


SeqKit 2 is now described in a new paper! It's great to see this great program growing. Inspiring! onlinelibrary.wiley.com/doi/full/10.10…
onlinelibrary.wiley.com
SeqKit2: A Swiss army knife for sequence and alignment processing
SeqKit2 has significantly expanded its capabilities, doubling the number of subcommands from 19 to 38, and adding support for three more compression file formats. This enhances its versatility and...

Did you know that fu-index allows you to extract lane and run information from Illumina datasets? This can be useful when splitying by run (e.g. when running DADA2 or prevent batch effect) telatin.github.io/seqfu2/utiliti…

Microsoft wants to buy us, but then they would make `SeqFu (work or school)` and `SeqFu (new)` available through their app store...

Complain all you want about peer review @FrontiersIn, but its current manifestation is the logical extension of a toxic, dysfunctional and irrational publishing culture that treats papers as currency and continues to propagate the lie that "peer reviewed" means "valid".
🥖 Freshly baked: Fasten by @lskatz was just published in @JOSS_TheOJ. Of course, it's a fast FASTQ manipulation toolkit, written in Rust. UNIX pipes provided. Bring your own dataset :)

Metagenome taxonomy profilers usually ignore unknown species. SingleM is an accurate profiler which doesn't, even detecting phyla with no MAGs. We applied it to 250,000 metagenomes- in environmental samples 75% lack a genome/MAG (abundance-weighted). A 🧵biorxiv.org/content/10.110…

Thanks! >1_1 frame=+0 start=0 tot=3 MERRYCHRISTMASANDAHAPPYNEWYEAR with SeqFu ORF telatin.github.io/seqfu2/utiliti…

ATGGAGAGGAGGTACTGCCACAGGATCAGCACCATGGCCAGCGCCAACGACGCCCACGCCCCCCCCTACAACGAGTGGTACGAGGCCAGG from everyone at @ensembl

Hey #Miniconda users, `conda-libmamaba-solver` is default solver since 23.10, so stop installing mamba (Or use #micromamba straight away! 😜)
Have a nice weekend, folks! Keep your #HPC clusters busy over the weekend with your #bioinformatics analyses 🙃

Thanks for the GitHub stars, I just realise they are slowly growing. Can we hit the 128 🌟 (2^7)? Thanks :)


A new #SeqFu release directly from the Gambia! Bugfix, slight improvement to fu-orf (1.20.1) And moved the building stack to nim 2.0 Unfortunately, bioconda recipe won't work on macOS for the latest version... you can get up to 1.20 on mac until this gets fixed

Also this year, we had the opportunity to share some #SeqFu stickers at #EBAME8 with @EvelienAdri, @HannahVPye and @telatin. Every single sticker is honoured...

Have a good day folks. Today I was wondering: are #bioinformaticians engaged in the @htmx_org framework hate/love/laser debate?


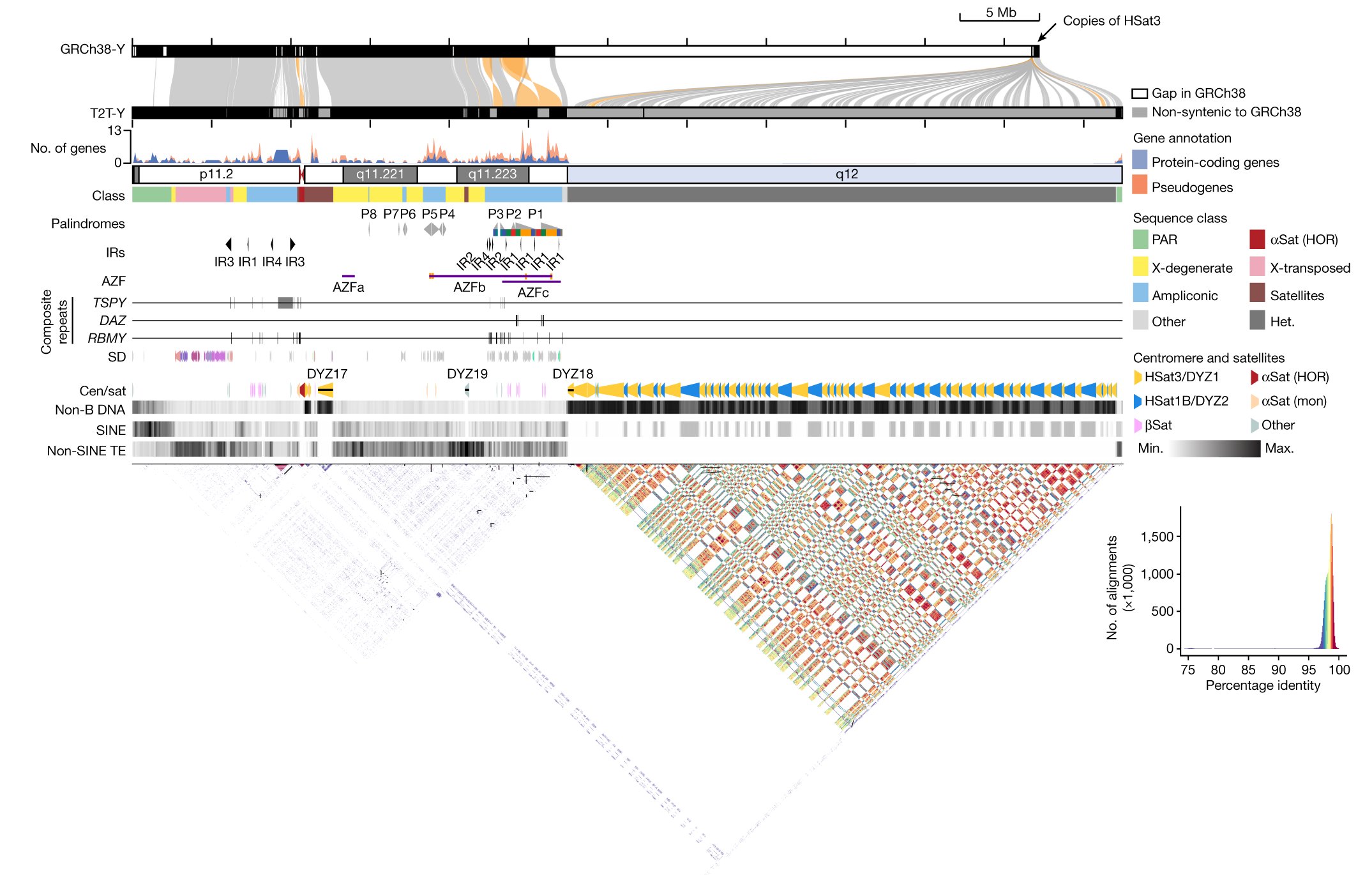
A very intriguing result in the new Y chromosome paper, one that you might miss unless you read the paper closely... 1/6 nature.com/articles/s4158…
































































































United States Trends
- 1. Josh Allen 40.2K posts
- 2. Texans 60.4K posts
- 3. Bills 152K posts
- 4. #FridayVibes 2,765 posts
- 5. Joe Brady 5,344 posts
- 6. #MissUniverse 472K posts
- 7. #MissUniverse 472K posts
- 8. Technotainment 19.8K posts
- 9. #Ashes2025 25.4K posts
- 10. Troy 12.3K posts
- 11. Anderson 28.2K posts
- 12. McDermott 4,770 posts
- 13. joon 13.2K posts
- 14. #StrayKids_DO_IT_OutNow 54.3K posts
- 15. OpenMind 52.3K posts
- 16. Beane 2,916 posts
- 17. FINAL DRAFT FINAL LOVE 1.31M posts
- 18. GM CT 24.5K posts
- 19. Maxey 15K posts
- 20. Shakir 5,705 posts





Something went wrong.
Something went wrong.