
Jim Shaw
@jim_elevator
Postdoc with Heng Li at Dana-Farber/Harvard Med School. Math PhD from @UofT with @YunWilliamYu. Working on methods for analyzing (metagenomic) sequencing data.
You might like











Stunning feat of applied sequencing bioinformatics. Congrats to the authors!!
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵 Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open. doi.org/10.1101/2024.0…

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵 Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open. doi.org/10.1101/2024.0…
skani v0.3.0 is released. github.com/bluenote-1577/… * 30-40% potential reduction in memory * Breaking changes to indexing and searching databases Calculate ANI for contigs, genomes. Search vs > 140k genomes: pre-indexed GTDB-R226 available for download.


I'm excited to share that I will soon join UCLA's Bioengineering department as an Assistant Professor. I am incredibly fortunate to have landed my dream job in this current atmosphere, and I would like to thank my mentors, colleagues, and friends for their support.


Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment. arxiv.org/abs/2506.12986


New life update! 🎆 🎓 This Fall, I will be joining the Department of Computer Science at Johns Hopkins University (@JHUCompSci) as an Assistant Professor, with an affiliation at the new Data Science and AI Institute (@HopkinsDSAI).


@csuhuangneng developed longcallR for joint SNP calling and phasing from long RNA-seq reads, AND for identifying allele-specific splicing/junctions (ASJ). Although ASJs of statistical significance are rare, a large fraction involve unannotated junctions. In Rust!

SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads biorxiv.org/content/10.110… #biorxiv_bioinfo
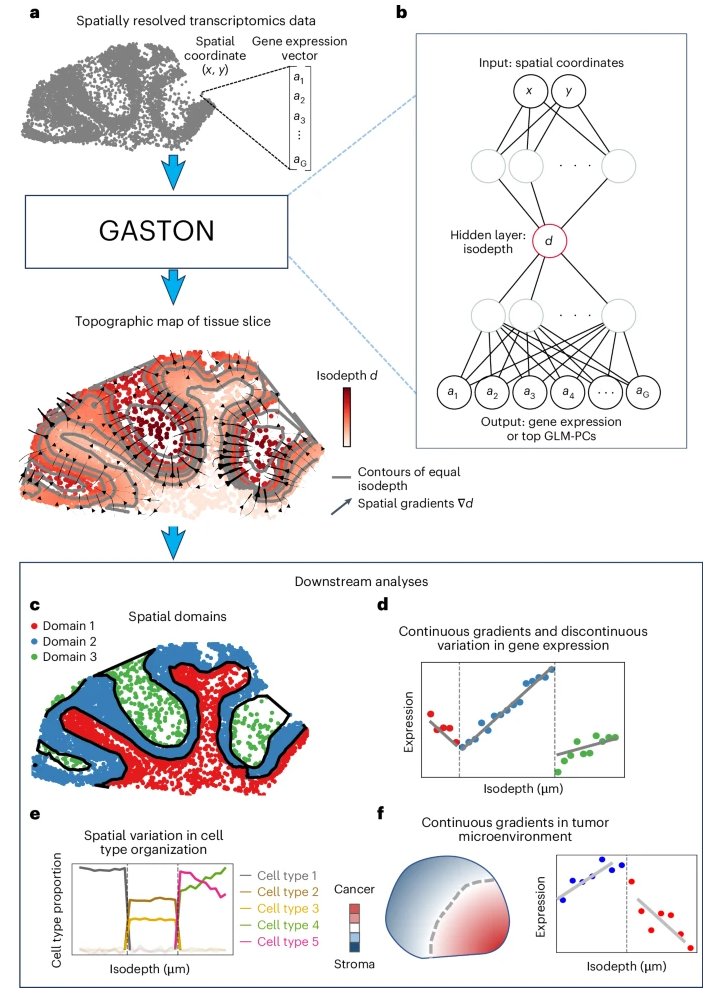
GASTON, our method to learn “topographic maps” of gene expression, is out now @naturemethods! IMO the coolest part is a new model of *spatial gradients in sparse data*. As is typical for bio papers, it’s buried in Methods, but see below for a quick outline on the math 👇

Gene expression topography analysis by GASTON portrays domain organization and spatial gradients of gene expression and cell type composition using spatially resolved transcriptomics data. @uthsavc @benjraphael @PrincetonCS nature.com/articles/s4159…

Tech Alert! 🚀🧬 We can now determine the sequence of DNA with non-canonical bases in a direct and high-throughput manner with Nanopore sequencing. Check out our preprint for details: biorxiv.org/content/10.110…
Hifiasm 0.21.0 has been released. It now has a beta module for direct assembly of ONT R10 simplex reads. Initial tests with regular simplex reads show very promising results! github.com/chhylp123/hifi…


Happy to see our method for T2T genome assembly published! It addresses an important limitation of string graph, that is, the contained reads. Led by @skamath5e "Telomere-to-telomere assembly by preserving contained reads" doi.org/10.1101/gr.279…

SPECIAL ISSUE! This month @genomeresearch publishes a diverse collection of research and review articles in a special issue highlighting advances in long-read sequencing applications in biology and medicine. tinyurl.com/Genome-Res-34-….

Just tried Sylph github.com/bluenote-1577/… by @jim_elevator. Processed >2000 samples in a few hours on google cloud powered by @TerraBioApp. Roughly 15min per sample. Briefly checked the abundances and they match pretty well with what we got using an independent workflow.

I gave a talk recently at Ben Langmead's group on my post on fast computation of random minimizers. Was super fun! Blogpost: curiouscoding.nl/posts/fast-min… Recording: curiouscoding.nl/talks/minimize…

A new paper from our amazing @sladky_on (jointly supervised with @VeselyPavel_mff) on super space-efficient indexing of arbitrary k-mer sets, introducing the Masked Burrows-Wheeler Transform (MBWT).
FroM Superstring to Indexing: a space-efficient index for unconstrained k-mer sets using the Masked Burrows-Wheeler Transform (MBWT) biorxiv.org/cgi/content/sh… #biorxiv_bioinfo

























































































United States Trends
- 1. Steelers 51.6K posts
- 2. Rodgers 21K posts
- 3. Chargers 35.9K posts
- 4. Tomlin 8,138 posts
- 5. Schumer 217K posts
- 6. #BoltUp 2,898 posts
- 7. Resign 102K posts
- 8. #TalusLabs N/A
- 9. Tim Kaine 17.8K posts
- 10. #HereWeGo 5,617 posts
- 11. Keenan Allen 4,725 posts
- 12. #RHOP 6,777 posts
- 13. Durbin 25.1K posts
- 14. Herbert 11.5K posts
- 15. #ITWelcomeToDerry 4,349 posts
- 16. Gavin Brindley N/A
- 17. Angus King 15.1K posts
- 18. Ladd 4,360 posts
- 19. 8 Democrats 8,520 posts
- 20. 8 Dems 6,621 posts
You might like
-
 Rayan Chikhi
Rayan Chikhi
@RayanChikhi -
 PM @[email protected] @pashadag.bsky.social
PM @[email protected] @pashadag.bsky.social
@pashadag -
 Sebastian Deorowicz
Sebastian Deorowicz
@sdeorowicz -
 Mikhail Karasikov
Mikhail Karasikov
@m_karasikov -
 Andrea Guarracino
Andrea Guarracino
@AndresGuarahino -
 Karel Břinda
Karel Břinda
@KarelBrinda -
 Ragnar {Groot Koerkamp} 🦋
Ragnar {Groot Koerkamp} 🦋
@curious_coding -
 Santiago Marco-Sola
Santiago Marco-Sola
@santiagomsola -
 Giulio Ermanno Pibiri
Giulio Ermanno Pibiri
@giulio_pibiri -
 Nae-Chyun Chen
Nae-Chyun Chen
@naechyun_chen -
 Massimiliano Rossi
Massimiliano Rossi
@maxrossi91
Something went wrong.
Something went wrong.